The identification of Trichoderma species is a difficult task. The description of a new species is a taxonomic act that can not be undone. Therefore, do not hesitate to contact the experts before proposing new Trichoderma species.
ICTT RECOMMENDATIONS RELATED TO TRICHODERMA DIVERSITY STUDIES
PRIMERS AND PCR CONDITIONS SUITABLE FOR TRICHODERMA DIVERSITY RESEARCH
MOLECULAR IDENTIFICATION PROTOCOL FOR TRICHODERMA
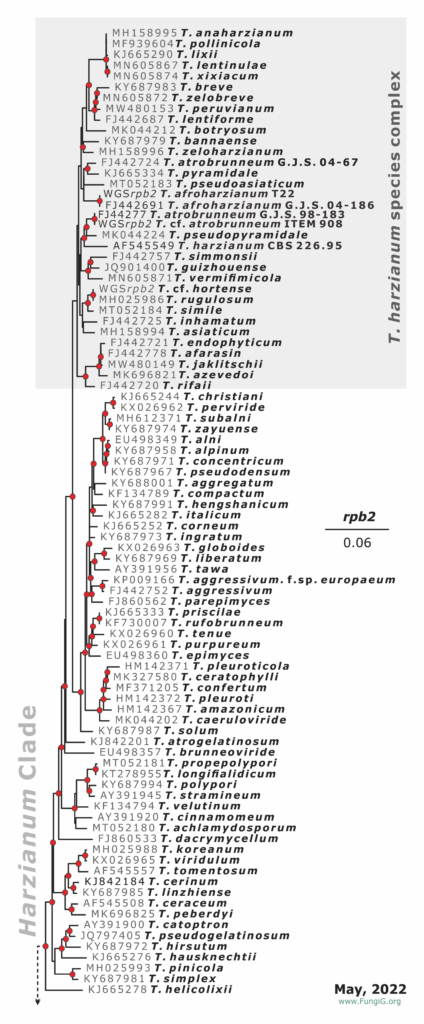
The most complete rpb2 Trichoderma tree with 391 RefSeq (May 10, 2022)
WARNING! The taxonomy of the Harzianum Clade, including the T. harzianum species complex sensu Chaverri et al. (2015), is challenging. The recognition of taxa in this group requires a unification of species criteria and additional DNA Barcoding loci. Please note that the application of the Genealogical Concordance Phylogenetic Species Recognition concept (Taylor et al., 2000) is complicated by the putative recombination between distantly related strains (Druzhinina et al., 2010). Therefore, species identification in this group requires a detailed analysis of single loci phylogenies for rpb, tef1, and preferably other marker genes (see below). Please do not hesitate to contact us!
Phylogeny of the Harzianum Clade based on the ML analysis of the partial sequence of the rpb2 gene as of May 2022. This phylogram is provided for reference only.
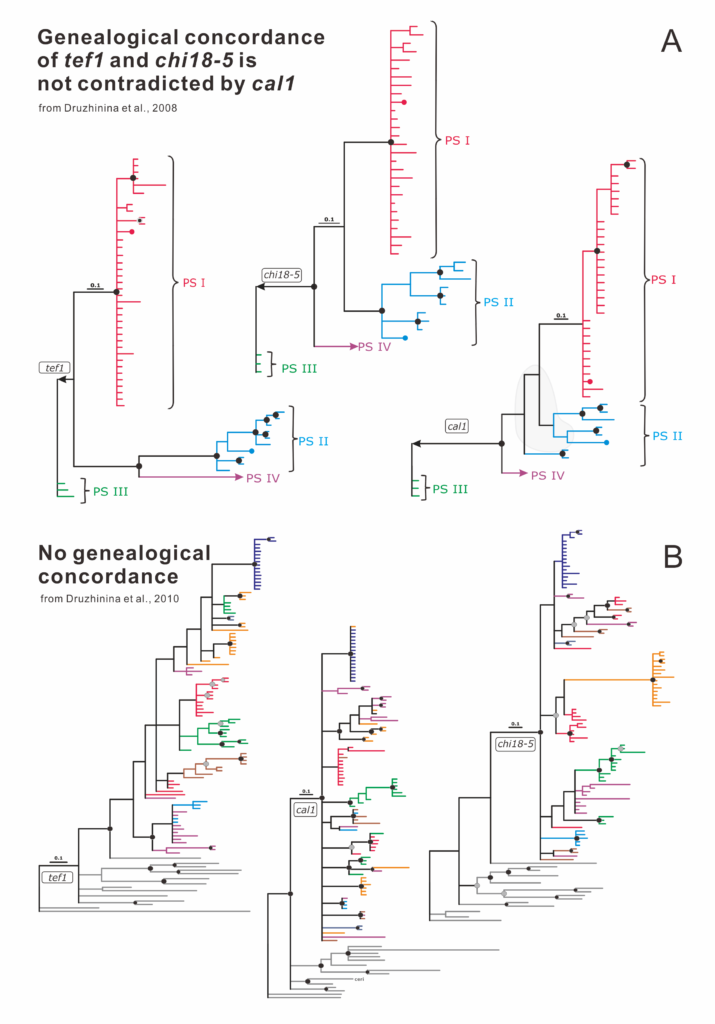
Examples of genealogical concordance in the genus Trichoderma
A. Phylogenetic trees taken from (Druzhinina et al. 2008) that describe phylogenetic concordance of the three loci (tef1, chi18-5, and cal1) in the Section Longibrachiatum. PS I–PS IV correspond to phylogenetic species. Colors indicate statistically supported clades of the concatenated phylogram of the three loci. See (Druzhinina et al. 2008) for details. B. Phylogenetic trees taken from (Druzhinina et al. 2010) describing the lack of phylogenetic concordance of the three loci (tef1, chi18–5 and cal1) in the Harzianum Clade. Colors indicate clades seen on the concatenated phylogram of the three loci. Modified from Cai and Druzhinina, 2021