The identification of Trichoderma species is a difficult task. It has been recently addressed by Cai and Druzhinina (2021) that developed the
MOLECULAR IDENTIFICATION PROTOCOL FOR A SINGLE TRICHODERMA ISOLATE
The following molecular identification protocol enables a user 1) to identify the genus Trichoderma, i.e. to exclude fungi other than Trichoderma, 2) to identify Trichoderma species, and 3) to verify the ambiguity of the identification. The protocol allows the recognition of a putative new species as a particular case of species identification.
All steps proposed below refer to the taxonomic limitations constraining the molecular diversity of the genus Trichoderma and recognized species that exist by July 2020.
A species of Trichoderma can be identified if its ITS sequence reaches at least one similarity value ≥76% to the sequences in the dataset attached to the protocol in Cai and Druzhinina (2021) and the two other DNA Barcoding markers are highly similar to the corresponding sequences of the reference strain of one species as rpb2 ≥ 99% AND tef1 ≥ 97%. These conditions can be shortened as the following sequence similarity standard:
Trichoderma[ITS76]~sp∃!(rpb299 ≅ tef197),
where Trichoderma means the genus Trichoderma, sp means a species, “~” indicates an agreement between ITS and other loci, “≅” refers to the concordance between rpb2 and tef1, and ∃! points to the uniqueness of this conditions (only one species can be identified). Subscripts show the similarity per each locus sufficient for the identification following the assumptions of the protocol below. The flowchart of the protocol is given in the Figure below.
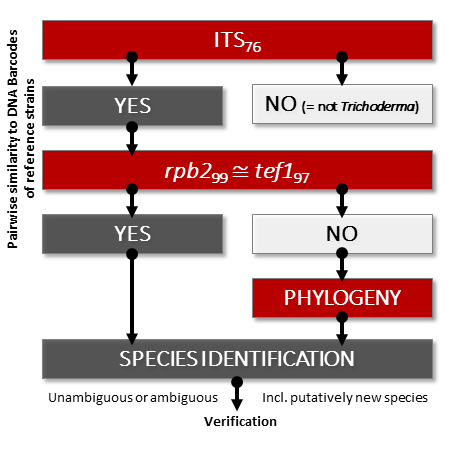
The flowchart of the molecular identification of Trichoderma based on three DNA Barcode sequences.
A species of Trichoderma can be identified if its ITS sequence reaches a similarity value ≥76% (ITS76) to the sequences in the dataset attached to the protocol and the two other DNA Barcoding markers are highly similar to the corresponding sequences of the reference strain of one species as rpb2≥99% AND tef1≥97% (rpb299≅ tef197), “≅” refers to the concordance between rpb2 and tef1. Cai and Druzhinina (2021)
The result of molecular identification requires biological verification (Lucking et al., 2020) and the consideration of the original taxonomic literature. The morphology and growth profile of the query strain should not contradict the published records for the identified species. It is recommended to compare the biogeography and occurrence records for the identified species with metadata for the query strain. The observed life cycle, ecology (habitat and interactions with other organisms), and ecophysiology of the query strain should be in agreement with the description of the identified species. For ambiguous cases, taxonomy experts can provide useful consulting.
The check-list for materials, tools and preparation steps.
- Isolate a single spore (asco – or conidiospore) culture of the putative Trichoderma sp. culture.
Note: although the fast growth on rich nutritional media, mycoparasitism, resistance to xenobiotics, and greenish conidiation are the characteristic features for most of the Trichoderma cultures, some species have hyaline conidia or do not produce them in vitro (appear white in culture), some are sensitive to fungicides, some do not parasitize on other fungi and / or have slow growth in vitro. Refer to the diversity of Trichoderma spp. phenotypes in monographs of Walter M. Jaklitsch (refs) or elsewhere.
- PCR amplify and sequence the three DNA Barcode loci: the complete fragment of ITS1 and 2 (including the 5.8S rRNA) of the rRNA gene cluster, and partial sequences of rpb2 and tef1 genes.
Note: PCR protocols including corresponding primer pairs are recommended in Table 3, the structure of the loci is shown in Figure below:
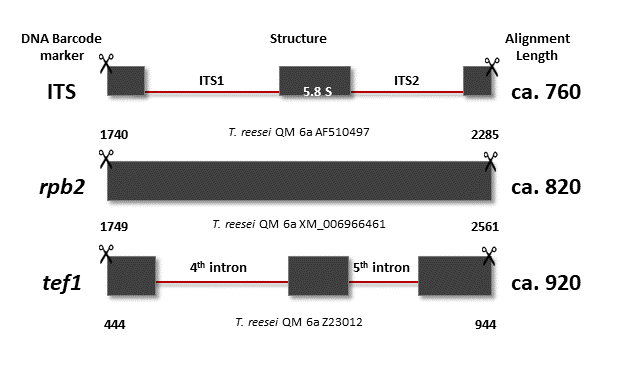
Numbers below each locus show the 5’ and 3’ positions on the trimmed fragment suitable for molecular identification using reference loci from T. reesei QM 6a (Druzhinina et al., 2005, 2010, Cai and Druzhinina, 2021)
Note: The quality of obtained sequences is crucially important for this protocol. No ambiguity in sequencing reaction is accepted. Ideally, sequences should be verified by sequencing from 3’ and 5’ ends.
- Connect to Internet.
- Trim the sequences. Use TrichoMARK 2020 available at www.trichokey.com or the reference datasets (Supplementary Dataset and www.trichoderma.info) and trim the length of the query sequences such that they correspond to the length of the reference DNA Barcode loci as shown on Figure 9.
Note: this step is compulsory for the molecular identification protocol. If online tools are not available, the sequences can be trimmed manually using Aliview (ref) or other sequence editors. The technical requirement to trim the sequences is explained in Kopchinskiy et al., 2005.
- Use a text editor (e.g. Notepad) and put your trimmed query DNA Barcode sequences in FASTA format and save the input files separately.
- Install Aliview (ref), IQ-TREE (ref), and FigTree (ref), or software with similar functions.
Step 1: ITS
Estimate the pairwise similarity between the ITS sequence of the query strain and the sequences given in the ITS56 dataset as described in Comment 1.
If the maximum similarity is ≥76% the query strain belongs to the genus Trichoderma spp. Continue to Step 2
If the maximum similarity is <76%, the query strain belonging to the genus other than Trichoderma. Identification of Trichoderma spp. is not possible
Step 2: rpb2 and tef1
For each locus (rpb2 and tef1, respectively), estimate the pairwise similarities between the query strain and the sequences of closely related reference strains as described in Comment 2.
If the condition ∃!(rpb299 ≅ tef197) is met, record identified species and continue to Step 4
If the condition ∃!(rpb299 ≅ tef197) is not met, continue to Step 3
Step 3: phylogenetic analyses of rpb2 and tef1
- Open rpb2 and tef1 (separately) alignments produced in Step 2 (see Comment 2).
- Record cases of short or missing sequences for reference strains (if any).
- Run phylogenetic analyses separately for rpb2 and tef1 sequences. Given the correct parameters selected, the maximum likelihood or Bayesian methods are recommended; however maximal parsimony is also suitable. See details in Materials and Methods.
- Visualize the tree files in Figtree (ref) and (optionally) export to a graphic software.
- On each tree, locate the query sequence and the most similar reference sequences; mark the pairwise similarities estimated in Step 2 (examples are shown on Figures 10 – 13).
- Interpret the concordance of rpb2 and tef1 phylograms considering the similarity values estimated in Step 2.
Note: the consideration of single loci phylograms of tef1 and rpb2 is required. The concatenated phylogram of the two loci is optional in addition to the analysis of single locus trees. For the interpretation of phylogenetic trees refer to Comment 3 and practical examples below.
Step 4: Validation of molecular identification
Note: For the validation of the molecular identification and assignment of ambiguity status, the literature on Trichoderma taxonomy should be studied. Table 2 of this study (also available at www.trichokey.com) provides supplementary information.
In some cases, results of phylogenetic analysis (Step 3) can be used for the validation of identification results (Comment 3).
Validation of species identification
If all of the following criteria are met:
- Identified species is represented by the complete set of DNA Barcodes (Table 2, www.trichokey.com; taxonomic literature).
- The identifiability of the species is not compromised by insufficient polymorphism of tef1 and rpb2, or other parameters (= there are no warnings in Table 2).
- The identified species was recognized based on the GCPSR concept.
The identification is unambiguous, precise and accurate
If any of the following criteria are met:
- The identified species is represented by the incomplete set of DNA Barcodes (see warnings in Table 2 or at www.trichokey.com).
- The identifiability of the species is compromised by low polymorphism of tef1 and rpb2, or the quality of the reference sequences is not sufficient (usually, too short) (see warnings in Table 2 or at www.trichokey.com).
- The identified species was recognized based on insufficient reference material or ambiguous species criteria.
The identification is ambiguous; the species name can be assigned as “confer” or “cf.” [= compare to] the most closely related species
Note: In this case, the most closely related species can be revealed based on the results of phylogenetic analysis (Step 3, Comment 3). The precise and accurate identification will usually require either taxonomic revision of reference materials, additional DNA sequencing or/and sampling.
Validation of the new species hypothesis
If all of the following criteria are met:
- The query strain belongs to the genus Trichoderma (meets Trichoderma[ITS76]standard)
- The query strain has unique sequences of rpb2 or tef1 (does not meet the sp∃!(rpb299 ≅ tef197) standard of being the known species because of the lower similarity of one or both loci)
- The existing closely related species have complete sets of reference DNA Barcodes
- The new species hypothesis is supported by the topology of both phylograms (rpb2 and tef1) and is not contradicted by other markers (GCPSR concept).
The new species hypothesis is unambiguous, precise and accurate. Record the results as “T. sp. strain ID” before the formal name is given.
Note: the formal taxonomic description of a new fungal species requires following the guideline of Seifert and Rossman (2010) including naming (see The Code), the deposition of the reference materials in public databases, microbiological investigation and imaging of microscopic features. It comprises the molecular evolutionary analysis (Comment 3), comparison of morphological, eco-physiological, and biogeographical characters between the query strain(s) and closely related taxa (ref).
If any of the following criteria met:
- The attribution of the query strain to the genus Trichoderma is ambiguous (does not meet Trichoderma[ITS76]standard, in particular if the similarity is <70%)
- The closely related species have incomplete sets of DNA Barcodes or the quality of the reference sequences is not satisfactory or related species were recognized based on insufficient DNA Barcoding material.
- The position of a new species is not supported by the topology of both phylograms (rpb2 and tef1) or is contradicted by other markers (GCPSR concept is not applicable).
The hypothesis of a new species remains ambiguous
Note: In this case, the species name can be assigned as “confer” or “cf.” [= compare to] the most closely related species which can be revealed based on the results of phylogenetic analysis (Step 3, Comment 3). The precise and accurate identification of a new species will usually require either taxonomic revision of reference materials, additional sequencing or/and sampling.
Step 5: Presentation of the identification result and data archiving
- Record the identification results. An example is given in Table 4.
- Archive your non-trimmed query DNA Barcode sequences along with their identification (FASTA format is suggested).
Comments:
Comment 1. Calculation of pairwise similarities between the query and reference sequences using ITS:
- Download the sequence ITS56 dataset from (Supplementary material Dataset of this study or www.trichokey.com) and open in the text editor. Add the query ITS sequence to the dataset.
- Insert the sequences in Aliview and use “Realign everything” option in “Align” menu.
- Check whether the length of the query sequence fits the ITS56 dataset. If not, the identification result will be ambiguous.
- Export the alignment as .fasta file and save it.
- Upload the exported .fasta file or paste the sequences to the input box of the online tool of ClustalOMEGA for pairwise similarity calculation (https://www.ebi.ac.uk/Tools/msa/clustalo/) or use other tools for pairwise sequence similarity calculation.
- Select the option of “DNA”, setup your parameters (“ClustalW” is recommended) and click the button of “submit”.
- Download the .pim file which contains the result of the pairwise similarity calculation, from the page of “results summary”.
- (Optional) A “guide tree” can also be obtained from the page of “results summary” and visualized in FigTree for your interest.
- Open the .pim file with Microsoft Excel or a text editor (.txt), search for the maximun similarity value(s) between your query sequence and the references. Make sure you have excluded the value showing the similarity to the query sequence itself (100%).
Note: The ITS56 dataset contains 56 selected reference ITS sequences that represent intra-generic polymorphism of the Trichoderma genus.
Comment 2. Manual calculation of pairwise similarities between the query and reference sequences using tef1 or rpb2:
- Submit the trimmed rpb2 sequence to TrichoBLAST (www.trichokey.com ) and detect the most closely related species.
- Use the most updated Table 2 (namely the latest updated version on www.trichokey.com) and taxonomic literature published after the release of this manual and compose the lists of the most closely related species, 6<N<10.
- Find the taxonomically confirmed reference strains (ex-type, type, vouchered, Table 2) for each species and retrieve rpb2 and tef1 sequences from public databases.
- Align and trim the sequences, and calculate pairwise sequence similarities as described in Comment 1.
Comment 3. Application of phylogenetic analysis in molecular identification and its use for the validation of identification results
Phylogenetic analysis can contribute to unambiguous or ambiguous identification of either a known species or a putative new species:
- Case 1: If the sequence similarity standard (no matter rpb2 and/or tef1) points to several species (e.g. T. cf. endophyticum CFAM-422, Tables 1 and 4), phylogenetic analysis of both loci will reveal the closest species and allow accurate but imprecise (ambiguous) identification as Trichoderma cf. [closest species]. However, this analysis will usually indicate a need for the taxonomic revision of the reference group. In this case, phylogeny is used as an identification step.
- Case 2: If the two loci point to different species (existing or putatively new), the results of the phylogenetic analysis can demonstrate that the loci are not concordant (e.g. T. sp. nov. NJAU 4742, Tables 1 and 4). In this case, and considering that only two markers are currently available, phylogeny is used as a validation step. With the introduction of genomic techniques in fungal taxonomy, such cases may be resolved by the application of phylogenomic analyses (refs).
- Case 3:
- Case 4: If a new species is found, phylogeny is a required part of the new species identification. In this case, the topologies of both phylograms are expected to be concordant and pairwise sequence similarities should support the unambiguous new species hypothesis.


